
Table of Contents
Thalassemia is a spectrum of genetic conditions in which one or more globin genes are defective, resulting in reduced or deficient synthesis of the related globin chains. Patients with minor genetic abnormalities are usually asymptomatic. Patients with more severe abnormalities have symptoms caused by one or more of the following: low formation of normal hemoglobin, synthesis of defective hemoglobin, imbalanced synthesis of α- and non-α-globin chains, and inefficient erythropoiesis.
The term thalassemia is derived from the Greek word thalassa, which means “from the sea.” Because of the significant prevalence of this illness in patients of Mediterranean origin, Whipple et.al., in 1932 named the disease ‘thalassemia’ or ‘Mediterranean anemia’. Several researchers identified the genetic cause for this anemia in the 1940s and were able to prove that individuals who were homozygous for this disorder (thalassemia major) had a severe outcome. The heterozygotes, on the other hand, were not just carriers but also had a lesser anemia (thalassemia minor).
Thalassemias are globin chain diseases characterized by a loss of synthesis of alpha or beta globin chains. Thalassemias are hemoglobin formation disorders. Inability in synthesis of either the alpha or beta chains inhibits the formation of normal adult hemoglobin, hemoglobin A (α2,β2), hemoglobin A2 (α2,δ2), and hemoglobin F (α2,γ2).
Mechanisms in α-Thalassemia
α-Thalassemias are conditions characterized by decreased production of α-globin chains, which are primarily caused by inherited α-globin gene deletion. In the normal diploid form, each copy of chromosome 16 includes two tightly connected α-globin genes, for a total of four α-globin genes. α-thalassemias of varying severity are induced by deletions that result in loss of one to four α-globin loci.
Clinical features
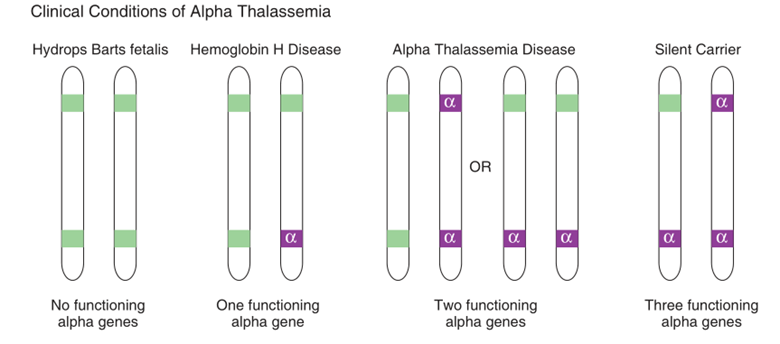
Alpha thalassemias are prevalent among Asian people (e.g., Thailand, Vietnam, Cambodia, Indonesia, and Laos). They have also been observed in Saudi and Filipino communities. Because each adult hemoglobin is dependent on the development of the alpha chain, the alpha chain is a fundamental building block in the formation of all normal adult physiological hemoglobins. The alpha chain is also essential for the formation of hemoglobin in the fetus; without it, no Hgb F is generated. The number of alpha genes removed determines one of four clinical stages of alpha thalassemia.
- Loss of four α-globin loci: Hydrops fetalis causes mortality in pregnancy or shortly after delivery. The majority of hemoglobin is Hb Barts, a tetramer formed of fetal hemoglobin (γ4), which has such a strong O2 demand that it transfers very little O2 to tissues.
- Loss of three α-globin loci: HbH disease is distinguished on the basis of β-4 hemoglobin (hemoglobin H, which is made up of a β-globin tetramer). Jaundice, splenomegaly, and mild microcytic hypochromic anemia appear in infants.
- Loss of two α-globin loci: α-thalassemia minor is characterized by microcytosis, hypochromasia, and moderate anemia.
- Loss of one α-globin locus: α-thalassemia minima, with very little clinical or laboratory indications.
Hb Bart Hydrops Fetalis Syndrome
Homozygous a0-thalassemia (– –/– –) is characterized by the lack of all alpha chain formation and typically leads in fatality in utero or shortly after childbirth. The fetus is highly anemic, which causes heart failure and edema in the fetal subcutaneous tissues (hydrops fetalis). The primary hemoglobin is Hb Bart (γ4), with traces of Hb Portland (ζ2γ2) and Hb H. Hb Bart has an extremely high affinity for oxygen hence it limits transport of oxygen to the tissues. The fetus is delivered prematurely and is frequently stillborn or dies shortly after delivery.
Hemoglobin H Disease
The most common cause of Hb H disease is the deletion of three a-globin genes, leaving just one α-globin gene to create a chain (– –/–α). Because of the predominance of the α0 gene haplotype (– –), this genetic anomaly is more frequent in Asians. In adults, it is characterized by the formation of extra unpaired β-chains, which form tetramers of Hb H. Hb Bart makes up 10% to 40% of the hemoglobin in newborns, with the rest being Hb F and Hb A. Following the γ to β switch, Hb H replaces most of the Hb Bart, resulting in Hb H ranging from 1% to 40%, with a little quantity of Hb A2, remnants of Hb Bart, and the remaining Hb A.
Hb H disease is distinguished by mild to moderate chronic hemolytic anemia, with hemoglobin concentrations average 7 to 10 g/dL and reticulocyte counts ranging from 5% to 10%, while clinical and laboratory results vary significantly. Erythroid overgrowth occurs in the bone marrow, and the spleen is frequently enlarged. Patients having deletional Hb H disease do not need transfusions on a frequent basis. Infection, pregnancy, or exposure to oxidative medicines, on the other hand, might produce a hemolytic crisis that would need temporary transfusions.
α-Thalassemia Minor
The most common cause of α-thalassemia minor is the deletion of two α-globin genes. It might be homozygous α+ (–α/–α) or heterozygous a0 (– –/αα). This illness is asymptomatic and is distinguished by moderate anemia (mean hemoglobin concentration is 12 to 13 g/dL) and microcytic, hypochromic RBCs. At birth, the percentage of Hb Bart is between 5% and 15%. Because the creation of α and β chains in adults is balanced, Hb H (β4) is rarely found. Homozygosity for non-deletional mutations in both α2-globin genes result in mild to severe hemolytic anemia, frequently accompanied by jaundice and hepatosplenomegaly.
Silent Carrier State
The silent carrier condition is caused mostly by the loss of one α-globin gene, leaving three functioning α-globin genes (–α/αα). The α/β chain ratio is approximately normal, and there are no hematologic abnormalities. There is a modest reduction in chain synthesis since one α-globin gene is missing. At birth, there is a minor excess of chains that form tetramers of Hb Bart (γ4) in the range of 1% to 2%. Other than genetic study, there is no reliable approach to determine silent carrier state.
De Novo and Acquired Forms of α-Thalassemia
There are two separate α-thalassemia syndromes that can be caused by acquired or de novo mutations:
- α-thalassemia associated with mental retardation and
- Hb H disease associated with MDS.
α-Thalassemia Associated with Mental Retardation
α-Thalassemia, also known as Hb H disease, can develop as a de novo anomaly in a rare disorder known as α-thalassemia with mental retardation syndrome (ATR). Affected patients suffer mental retardation and a variety of other developmental problems in combination with a nontraditionally inherited α-thalassemia trait or Hb H disease. De novo deletions affecting the whole α-globin gene cluster and neighboring DNA at the tip of chromosome 16 develop in certain cases, resulting in the so-called ATR-16 syndrome.
Acquired Hb H Disease Associated with Myelodysplastic Syndrome or ATMDS
Hb H illness has been observed on rare occasions along the course of several kinds of MDS and in individuals with other hematologic malignancies. The condition often affects elderly males over the age of 60. The degree of globin chain synthesis imbalance and α-globin mRNA insufficiency in impacted individuals’ erythroid cells is larger than that reported in the hereditary form of Hb H illness.
Laboratory Diagnosis of α-Thalassemia
Complete Blood Count with Peripheral Blood Film Review
Even though majority of thalassemias cause microcytic and hypochromic anemia, laboratory findings can range from borderline abnormal to severely abnormal, depending on the type and number of globin gene mutations. The hemoglobin and hematocrit are reduced, but the RBC count might be abnormally high in comparison to the degree of anemia, resulting in very low MCV and mean cell hemoglobin (MCH). The concentration of mean cell hemoglobin (MCHC) is also lowered.
α-Thalassemia minima and minor are usually found to have no laboratory findings. α-Thalassemia minor can cause microcytic anemia.
HbH disease: moderately severe microcytic hypochromic anemia, with:
- Hemoglobin concentrations from 3-11 g/dL
- MCV from 50-60 fL
- Peripheral blood film with target cells, red cell fragments, tear drop red cells, and basophilic stippling
- Unconjugated hyperbilirubinemia
- Elevated LDH
- Decreased haptoglobin
- HbH (usually 1-40% of total hemoglobin), identified as a characteristic fast migrating species on Hb electrophoresis
- Heinz bodies, detected with vital stains, due to precipitation of HbH in red cells
- Decreased HbA2 (1-2% of total Hb)
Hydrops fetalis: severe anemia (hemoglobin concentrations of 3-8 g/dL), with:
- Bizarre microcytic, hypochromic red cells and nucleated red cells apparent on blood films.
- Hemoglobin electrophoresis reveals Hb Barts (70-100%).
Reticulocyte Count
The reticulocyte count is increased, suggesting that the bone marrow is responsive toward a hemolytic event. The usual reticulocyte count in Hb H illness is 5% to 10%. Minimal reticulocytosis suggests poor erythropoiesis.
Supravital Staining
To elicit precipitation of the intrinsically unstable Hb H in a-thalassemia minor, Hb H disease, and silent carrier a-thalassemia, brilliant cresyl blue or new methylene blue stain may be used. Hb H inclusions (denatured b-globin chains) are microscopic, numerous, irregularly shaped greenish-blue entities that are consistently dispersed throughout the RBC. They create a pitted texture on the RBCs that looks like a golf ball or a raspberry.
Assessment of Normal and Variant Hemoglobins
Hemoglobin electrophoresis, cation-exchange high-performance liquid chromatography (HPLC), and capillary zone electrophoresis (CZE) are the primary clinical laboratory procedures used to detect and quantify normal and variant hemoglobins. Each approach has benefits and disadvantages, and no method can detect and measure all hemoglobins. To discover particular mutations in globin genes and determine the kind of thalassemia or hemoglobinopathy, molecular genetic testing is necessary.
Molecular Genetic Testing
For HBA1 or HBA2 gene mutations, PCR-based targeted mutation analysis may be used to identify the seven most prevalent deletional mutations. This technique identifies around 90% of all alleles, however the detection rate varies depending on the method. If the PCR based screening does not cover the testing, DNA sequencing of the HBA1 and HBA2 genes, as well as deletion/duplication analyses, can be carried out.
Alkali denaturation test
This test is based on the information that carriers having hypochromic RBCs, which results in lower osmotic fragility. For 5 minutes, a sample of anticoagulated blood is incubated in 0.375 % saline. Normal RBCs will lyse and the solution will clear because the solution is hypotonic. Patients with thalassemia, on the other hand, have hypochromic RBCs that will not lyse in 0.375 % saline, leaving the solution turbid. This test is not specific for thalassemia and will detect hypochromia caused by any condition, including iron deficiency anemia.
Important differential diagnosis
Thalassemia minor has microcytic, hypochromic RBCs that must be discriminated from iron deficiency anemia and other microcytic, hypochromic anemias. Microcytic, hypochromic anemias have a minimal differential diagnosis. Iron studies are essential for this differentiation. It is critical to distinguish between thalassemia minor and iron deficiency in order to prevent unwanted testing and treatments.
References:
- Rodak’s Hematology Clinical Principles and Applications, 5th Edition
- Hematology in Practice by Betty Ciesla,
- High-Yield Pathology, Hematopathology, Elsevier Saunders
- Hematology Basic Principles and Practice, 6th Edition, Elsevier Saunders pub.
- Clinical Laboratory Hematology, 3rd Edition, Pearson pub.