
Table of Contents
Arbitrarily, the terms karyogram, karyotype and ideogram have often been used. The term karyogram should be applied to a systematic sequence of chromosomes prepared either by drawing, digitizing, or extending photography, indicating that the chromosomes of a single cell will describe an individual’s or even a species’ chromosomes. To identify the regular or irregular, colonial or inherited, chromosomal complement of an individual, tissue or cell line, the term karyotype should be used. It is suggestive of using the term ideogram be reserved for the diagrammatic representation of a karyotype.
Chromosome Non-banding Techniques
The autosomal chromosomes are numbered from 1 to 22 based on descending order of their length. When using non banding simple staining techniques the chromosomes they can be distinguished into 7 groups based on their order of size and position of centromere.
Different parameters are used to differentiate the non-banding chromosome by International systems for Human Cytogenomic Nomenclature (ISCN 2016):
- The length of each chromosomes, expressed as a percentage of the total length of a normal haploid set, i.e., the sum of the lengths of the 22 autosomes and of the X chromosome;
- The arm ratio of the chromosomes, expressed as the length of the longer arm relative to the shorter one;
- The centromeric index, expressed as the ratio of the length of the shorter arm to the whole length of the chromosome.
| Group | Chromosomes | Morphology |
| Group A | 1,2,3 | Large metacentric chromosomes readily distinguished from each other by size and centromere position. |
| Group B | 4,5 | Large submetacentric chromosomes |
| Group C | 6,7,8,9,10,11,12,X | Medium sized metacentric or submetacentric chromosomes. The X chromosome reselbles the longer chromosomes with satellites. |
| Group D | 13,14,15 | Medium sized acrocentric chromosomes with satellites. |
| Group E | 16,17,18 | Relatively short metacentric or submetacentric chromosomes. |
| Group F | 19,20 | Short metacentric chromosomes. |
| Group G | 21,22 | Short acrocentric chromosomes with satellites. The Y chromosome bears no satellites. |
Banding Techniques
A band is a portion of a chromosome that appears darker or brighter than its neighboring segments using one or more banding techniques. Bands that stain darkly using one form can only stain slightly using another. There are no “interbands” and the chromosomes are visualized as a constant sequence of light and dark bands. The Giemsa dye mixture is commonly used as the staining agent in techniques that show an almost similar pattern of dark and light bands around the chromosomes. G-staining procedures and the resulting G-bands are the terms used to describe these techniques.
Code to describe banding techniques
Q = Q‐bands
QF = Q‐bands by fluorescence
QFQ = Q‐bands by fluorescence using quinacrine
QFh = Q‐bands by fluorescence using hoechst 33258
G = G‐bands
GT = G‐bands by trypsin
GTG = G‐bands by trypsin using Giemsa
GTL = G‐bands using Leishman
GTW = G‐bands using Wright stain
GPG = G‐bands using pancreatin and Giemsa stain
GUG = G‐bands using urea and Giemsa stain
GAG = G‐bands by acetic saline using Giemsa
C = C‐bands
CB = C‐bands by barium hydroxide
CBG = C‐bands by barium hydroxide using Giemsa
R = R‐bandsc
RF = R‐bands by fluorescence
RFA = R‐bands by fluorescence using Acridine orange
RH = R‐bands by heating
RHG = R‐bands by heating using Giemsa
RB = R‐bands by BrdU
RBG = r‐bands by BrdU using Giemsa
RBR = r‐bands by BrdU using acridine orange
T = T‐bands
TH = T‐bands by heating
THG = T bands by heating with Giemsa
THA = t‐bands by heating with Acridine orange
Giemsa banding (GTG, GTW, GAG, GTL)
The introduction of Giemsa banding (G‐bands) in 1971 by Sumner et al. was another major advance in the field of cytogenetics. It eliminated the need for an expensive fluorescence photomicroscope and provided permanently stained slides with very high‐resolution bands. Today, Giemsa banding (or similar Romanowsky dyes) is the most widely utilized staining technique for chromosome analysis.
In human chromosomes, the 30‐nm fiber of DNA forms loops of approximately 75–100 kb, which are tethered or anchored at their bases to form what is known as a chromosome scaffold. This structure can be seen by removing most histones and non-histones from the chromosomes and surface‐spreading them appropriately for electron microscopy. The residual structure appears as a scaffold, with numerous and extensive loops of DNA radiating from a coarsely fibrous structure that resembles a metaphase chromosome. The final packaging of the 30‐nm fiber of DNA into chromosomes results in a 10,000‐fold reduction in DNA length. Fixation in 3 : 1 methanol‐acetic acid is an important preliminary step for G‐banding. Fixation extracts a portion of all histones, especially H1, and a group of non-histones in the 50,000‐ to 70,000‐dalton (Da) range, but it is far from a complete removal of proteins. The crosslinking data indicated that the conformation of chromosomal proteins in relation to DNA strongly influences banding.
Giemsa stain is a complex mixture of dyes that may vary in concentration, purity, and ratio. The main components are the basic aminophenothiazine dyes – azure A, azure B, azure C, thionin, and methylene blue‐and the acidic dye, eosin. The thiazin dyes vary in the number of methyl groups attached to a core of two benzene rings bound together by nitrogen and a sulfur atom. Several studies have been done concerning the band‐producing ability of the individual components of Giemsa stain. Methylene blue or any of the azures alone produces banding.
Chromatin is divided into two main groups: heterochromatin and euchromatin. Constitutive heterochromatin is highly condensed, very repetitive, and transcriptionally inactive during interphase, in order for gene transcription to occur. One type of euchromatin, facultative heterochromatin, behaves like heterochromatin in a developmentally controlled manner, being transcriptionally inactive, late replicating, and condensed during interphase. The inactive X chromosome in the somatic cells of female mammals is an example of facultative heterochromatin.
G banding requires methanol/acetic acid‐fixed cells spread onto slides; cytoplasmic background interferes with good G‐bands, so proper cell dilution and spreading is essential. Slides are sufficiently dried (for 2–4 days at room temperature), or baked at low temperatures (e.g., 60 °C) for 2–18 hours, or high temperatures (e.g., 90–95 °C) for 20–60 minutes to dehydrate them (see Table 6.4). Slides that are insufficiently “aged” by natural means or by heating will not respond at all to the trypsin, no matter how long it is applied, and the chromosomes will appear unbanded.
Trypsin exposure depends on its brand, concentration, and temperature, but may also be affected by preparation and working conditions. Recrystallized forms have a higher level of activity, thereby requiring less time or a lower concentration. A stock trypsin solution can be prepared in advance, and small aliquots can be frozen until needed. At lower temperatures, the working solution has decreased activity. Higher pH (approximately 8) and higher temperature increase the enzymatic action of the trypsin and may be useful with cells that appear to be resistant to standard concentrations. Exposing slides to trypsin in a vertical fashion (rather than horizontal) may enhance its activity slightly.
A fetal bovine serum rinse is helpful in stopping the action of the trypsin. Serum contains alpha‐1‐antitrypsin, which inhibits the trypsin by complexing it with the protein in the serum. Other rinses that are commonly used include pH 8.0 buffer, 0.85% (normal) saline, Hanks’ balanced salt solution (HBSS), and 70% ethanol.
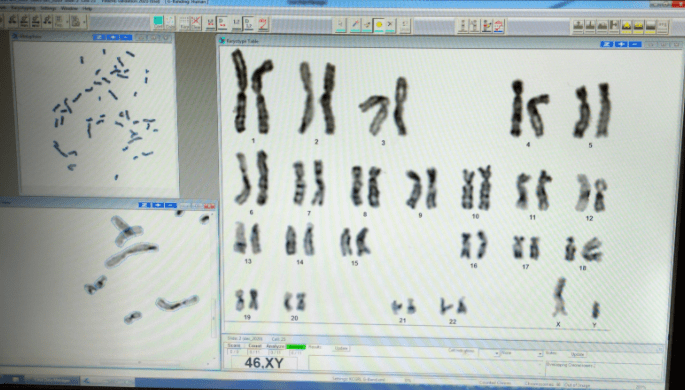
Determination of G‐banded chromosome resolution
As a rule, the better the banding resolution is, the better will be the chance of finding a small chromosome abnormality. This rule is more reliable for deletions and duplications than for centromere displacement (inversions, neocentromeres) because the constriction of the centromere becomes more difficult to visualize as the chromosomes become longer and thinner. However, there is a need to quantify the banding resolution of a given cell (on the analysis sheet) or chromosome study (on the final report) so that the limits of resolution can be inferred. There are several methods for determination of band levels.
Vancouver method
The Vancouver method involves counting bands in segments for some chromosomes (e.g., chromosomes 1, 11, and 12) and all bands in others (chromosomes 10 and X). Count only G‐positive bands and do not count the centromere. Total the bands then look at a chart that has increments of 50 bands at the lower range and 100 bands at the higher end to determine haploid band resolution.
| Chromosome | Resolution 350 | Resolution 450 | Resolution 550 | Resolution 850 |
| 1p31-p32 | 1 | 1 | 3 | 3 |
| 10 | 5 | 5 | 12 | 19 |
| 11p | 2 | 2 | 5 | 6 |
| 12q | 4 | 5 | 8 | 14 |
| X | 6 | 8 | 12 | 18 |
| Total | 18 | 21 | 40 | 60 |
Johnson and Stallard method
Johnson and Stallard used a method that quantifies the number of light and dark G‐bands present on one chromosome 10. Some laboratories account for a difference in resolution between the two chromosome 10 homologues in a cell by counting both homologues and dividing the total by 2 before using the haploid parameters.
| No of bands present | Haploid banding level |
| 12 | 375 |
| 13-14 | 400 |
| 15-16 | 425 |
| 17-18 | 450 |
| 19-21 | 475 |
| 22-23 | 500 |
| 24-25 | 525 |
| 26-28 | 550 |
| 29 | 575 |
| 30 | 600 |
| 31 | 625 |
| 32 | 650 |
| 33 | 675 |
| 34 | 700 |
| 35 | 725 |
| 36 | 750 |
| 37 | 775 |
| 38 | 800 |
| 39 | 825 |
| 40-41 | 850 |
Welborn method
The Welborn method involves counting all dark and light bands on one homologue each of chromosomes 1 and 2, counting the centromere as a band in each arm. Total the bands from both chromosomes and then multiply by 6 to get the haploid band resolution, since chromosomes 1 and 2 represent 1/6 of the genome.
References:
- The AGT cytogenetics laboratory manual
- An International systems for human cytogenomic nomenclature (ISCN 2016)