
Table of Contents
Cystic Fibrosis (CF) is a complex multi-system disease caused by mutations in the CFTR gene. It predominantly affects children, but about 41% of patients are adults, with 13% over 30 years old. Genetic testing and advancements in therapies are pivotal for diagnosis and treatment.
Key CFTR Gene Facts
- Gene Product: CFTR (cystic fibrosis transmembrane conductance regulator)
- Locus: 7q31.2
- Gene Structure: ~ 250,000 bp; 27 exons.
- mRNA: transcript for the gene is 6129 bp
- Coding Sequence (CDS): 4443 bp
- CFTR protein is 1480 Amino Acid; molecular weight of 168,173 Da
CFTR Gene Mutations and Mechanisms
The CFTR protein is made up of five domains: 2 membrane-spanning domains (MSD1 & MSD2) that make up the chloride ion channel, 2 nucleotide-binding domains (NBD1 & NBD2) that bind and hydrolyze ATP, and 1 regulatory (R) domain.
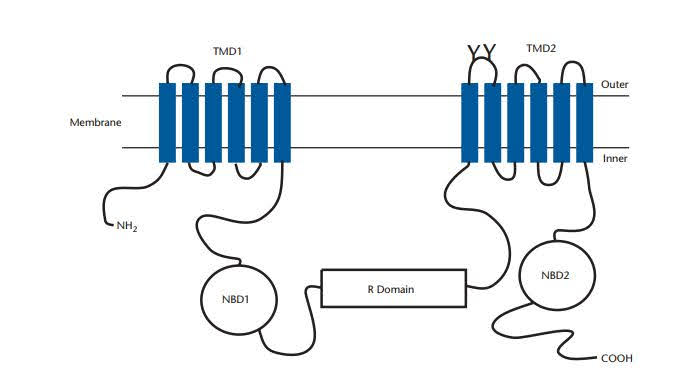
Six groups have been defined for CFTR gene mutations that influence development, different domains, and various functions. Class I (production) mutations truncate the protein, resulting in no active CFTR being made. Class II (processing) mutations are nontruncating and result in lower CFTR activity than natural. Class III (channel gating) mutations reduce chloride transfer by affecting the nucleotide binding domains (NBD1, NBD2).
The NBD and R domains are needed for the CFTR channel to open and close. Class IV (conductance) mutations change amino acids in transmembrane domains that make up the channel’s pore. Downregulation (Class C) mutations reduce the amount of functional CFTR produced. CFTR’s regulatory roles are affected by Class VI (regulatory) mutations. In general, CF cases in groups I, II, and III account for more than 85% of all cases.
The homozygous DF508 genotype is normally more serious than the other CFTR gene mutations. The severity of lung dysfunction varies among DF508 homozygotes, varying from moderate to extreme. The absence of a substantial connection between CFTR genotypes and phenotypes means that the phenotypic outcome is determined by other genes (modifier genes) and/or environmental factors.
Impact of Cystic Fibrosis on the Body
Understanding CF’s impact reveals its effects on multiple organs, especially those involving exocrine glands including the lungs, pancreas, and other organs. By comparing the functioning of these organs in a healthy individual to those affected by CF, we can gain insight into the disease’s mechanisms. While chronic bronchial infections and gradual lung decline are among the most severe health complications associated with CF, this multi-system disease affects various organ systems, particularly the exocrine glands.
In contrast to endocrine glands (such as the pituitary, thyroid, and adrenal glands) that release hormones directly into the bloodstream, exocrine glands release enzymes and metabolites into ducts that either exit the body or reach empty organs like the intestine. This fundamental distinction underscores the significance of understanding the natural functioning of these glands in order to comprehend the impact of CF on the body.
Lungs and the Respiratory System
‘Clubbing‘ an enlargement or rounding of the tips of the fingers and toes that occurs in almost all individuals with CF as a result of lung disease. Substances produced in response to lung infections tend to penetrate the bloodstream and promote the growth of soft tissue at the base of the fingernails and toenails in some way.
Pancreas and the Digestive System
Thick mucus can clog the pancreatic ducts in the majority of people with CF, particularly the smaller ones. Cysts and ultimately scar tissue develop as a result of the mucous plugs and the backup of enzymes in the pancreas. As a result, pancreatic enzymes are not able to enter the small intestine, allowing proteins and fats to pass through the body undigested.
Cystic Fibrosis affects on sweat glands
Sweat is made up of a dilute solution of chemical electrolytes, mostly sodium and chloride, with minor concentrations of calcium and potassium. While sweating excessive amounts of salt is rarely life-threatening, excessive electrolyte loss can be dangerous in hot weather or when a person with CF engages in strenuous exercise or develops a fever. People with CF are more likely to get dehydrated or suffer from heat stroke than healthy people. People with CF should drink electrolyte solutions or eat salty foods to compensate for the salt imbalance, particularly if they have a fever or are exercising vigorously.
Reproductive Systems
The vas deferens is completely absent in most men with CF, a syndrome known as congenital bilateral absence of the vas deferens (CBAVD). Women with CF go through normal sexual development and growth, but their sexual maturation can start slower or take longer than healthy women. Women with CF can have regular sex lives, and the majority of them can become pregnant, but they should weigh the risks carefully before choosing to have children. People with CF have a higher risk of spontaneous abortions, premature pregnancies, and stillbirths, which is mostly due to their respiratory issues.
| Organ/System | Impact of Cystic Fibrosis |
| Lungs and Respiratory System | -‘Clubbing’ of fingers and toes due to lung disease -Substances produced during lung infections promote soft tissue growth at the base of nails |
| Pancreas and Digestive System | -Thick mucus blocks pancreatic ducts, leading to cysts and scar tissue formation -Backup of enzymes in the pancreas prevents pancreatic enzymes from entering the small intestine -Undigested proteins and fats pass through the body |
| Sweat Glands | -Excessive electrolyte loss through sweat, primarily sodium and chloride -Increased risk of dehydration and heat stroke in hot weather or during physical activity -Recommended intake of electrolyte solutions or salty foods to compensate for salt imbalance |
| Reproductive Systems | -Absence of vas deferens in most men with CF (congenital bilateral absence of the vas deferens, CBAVD) -Normal sexual development and growth in women with CF, but slower maturation compared to healthy women -Increased risk of spontaneous abortions, premature pregnancies, and stillbirths, primarily due to respiratory issues |
Treatment Options for Cystic Fibrosis (CF)
Managing Cystic Fibrosis (CF) requires a combination of therapies to address the respiratory, digestive, and systemic effects of the disease. With ongoing advancements, patients today have access to improved treatments that enhance their quality of life. Here’s an overview of the most effective treatment options:
1. Airway Clearance
CF patients often face mucus buildup in the lungs, leading to respiratory issues. To help clear the airways, patients use techniques like:
- Chest Physiotherapy (CPT): Manual methods or vibrating vests loosen mucus.
- PEP Devices: Devices like the Flutter help expel mucus.
- Inhaled Medications: Medications like hypertonic saline, albuterol, and dornase alfa improve lung function by thinning mucus and easing breathing.
2. CFTR Modulators
CFTR modulator therapies target the CFTR protein defect, offering a breakthrough in treatment. These medications help improve chloride ion transport and function at the cellular level:
- Ivacaftor (Kalydeco): Helps restore function for certain CFTR mutations.
- Orkambi & Symdeko: These combination therapies target the F508del mutation, improving overall lung function.
- Trikafta: A triple combination therapy significantly enhances CFTR function, particularly in patients with the F508del mutation.
3. Gene Therapy & Innovative Treatments
Gene therapy is a promising avenue to correct the root cause of CF. CRISPR gene editing aims to correct mutations at the DNA level, while RNA-based therapies could adjust CFTR protein production, offering potential long-term solutions.
4. Infection Management
Frequent lung infections are common in CF, and managing them is essential. Patients use:
- Antibiotics: Oral antibiotics like azithromycin and tobramycin (inhaled) are used to target infections.
- Steroids: Medications like prednisone help reduce inflammation and improve breathing.
5. Nutritional Support
Due to digestive issues, CF patients need specialized care:
- Pancreatic Enzyme Replacement Therapy (PERT): Helps with digestion by providing missing enzymes.
- Vitamin Supplements: CF patients often require additional fat-soluble vitamins (A, D, E, K).
- High-Calorie Diet: A nutrient-rich diet supports weight maintenance and overall health.
6. Lung and Pancreas Transplantation
For severe cases, organ transplantation may be necessary:
- Lung Transplant: A lung transplant can provide a life-extending solution for advanced lung disease.
- Pancreatic Transplant: This helps restore enzyme production, benefiting patients with pancreatic insufficiency.
7. Managing CF-related Diabetes (CFRD)
Patients with CF may develop CFRD, a type of diabetes related to the disease:
- Insulin Therapy: Regular insulin treatment helps manage blood sugar levels.
- Monitoring: Frequent blood sugar checks are crucial to prevent complications.
8. Psychological and Emotional Support
Living with CF can be challenging, and mental health support is vital. Counseling, support groups, and stress management techniques help patients cope with the emotional toll of the disease.
References:
- Nussbaum, R. L., McInnes, R. R., & Willard, H. F. (2016). THOMPSON & THOMPSON GENETICS IN MEDICINE, EIGHTH EDITION. In Elsevier (8th ed., Vol. 6).
- Cystic Fibrosis Foundation