
Table of Contents
Primary immune thrombocytopenia, commonly known as idiopathic thrombocytopenic purpura(ITP), is an inflammatory bleeding disorder that affects both adults and infants. It was previously thought to be an autoantibody condition in which the reticuloendothelial system prematurely destroyed platelets that had been opsonized with antiplatelet antibody. Recent studies have revealed that there is a considerable underproduction of platelets in some cases, resulting to the thrombocytopenia seen in this condition.
Most people have no medical issues, however there could be bleeding. ITP has an unpredictable clinical course. Till now, treatments has mostly focused on minimizing platelet destruction through immunosuppression. Nonetheless, new thrombopoietin receptor agonists have been developed to promote platelet formation in the bone marrow. Due to immunosuppression, it is anticipated that these recent personalized therapies will be linked to a lower risk than conventional therapy.
Clinical features
The two main types of ITP are distinct in terms of their underlying etiology and symptoms.
Acute ITP
This is usually a pediatric disease, however an adult-related disorder does occur on sometimes. In a commonly normal child, the condition is defined by the sudden appearance of edema, petechiae, and, on rare occasions, mucosal bleeding (e.g., epistaxis). The most common hematologic sign is thrombocytopenia, which usually appears one to three weeks after an illness.
The most common type of outbreak is a non-specific upper respiratory tract or viral infection of the gastrointestinal tract, while acute ITP can also occur with rubella, measels (rubeola), chickenpox, or other viral infections that might precede live virus immunization. Acute ITP is estimated to affect 4 out of every 100,000 newborns, with a peak frequency in children aged 2 to 5 years. There is no gender preference. Thrombocytopenia persists for 6 months or more in approximately 10% to 15% of children who were initially diagnosed with acute ITP, and these children are reclassified as having chronic ITPs.
The finding that acute ITP frequently occurs in accordance with a viral disease suggests that certain children develop antibodies and immune complexes against viral antigens, and that attachment of these antibodies or immune complexes to the platelet surface can result in platelet destruction. Most patients with acute ITP recover in around 3 weeks with or without treatment, but others can take up to 6 months. Recurring episodes of severe ITP are uncommon in children who have recovered completely from the initial episode. Most people with acute ITP have mild symptoms and do not require treatment. However, serious conditions can be managed further, the most important beneficial approaches includes intravenous immunoglobulin (IVIG), platelet transfusions, and splenectomy (or any variation of them).
Chronic ITP
This disorder can affect people of any age, however the majority of cases occur in those between 20 to 50. Females outnumber men 2:1 to 3:1, with females being more prevalent between the ages of 20 and 40. Chronic ITP often progresses slowly, with platelet counts decreasing on a variable and frequently regular basis over long periods of time. The most common symptoms are menorrhagia, recurring epistaxis, and simple swelling (ecchymosis).
Platelet destruction in chronic ITP is the consequence of an immunological cycle. As a result of the implicated antibodies binding to platelets, reticuloendothelial cells, mostly in the spleen, start by removing the antibody-labeled platelets from circulation. Autoantibodies that recognize platelet surface glycoproteins such as glycoprotein IIb (GP IIb) and GP IIIa (aIIb/b3), as well as GP Ia/IIa, can be found in 50 to 60% of ITP patients. Because megakaryocytes frequently generate GP IIb / IIIa and GP Ib / IX on their membranes, these cells are obvious targets for the antibodies. Platelet turnover studies revealed that platelet development is affected during ITP. Overall, the platelet life cycle is lowered from 7 – 10 days to a few hours, and the rate at which platelets are removed form circulation is thought to be involved cause of thrombocytopenia.
When plasma from an ITP patient is administered into the circulation of a healthy receiver, the recipient develops thrombocytopenia. The plasma-producing agent for thrombocytopenia in ITP patients is an immunoglobulin G (IgG) antibody that may be recovered from serum by adsorption of typical human platelets. Furthermore, in vitro cytotoxic T cell-mediated platelet destruction is also seen using CD3, CD8 cells from active chronic ITP patients, although the in vivo significance of this mechanism has not been confirmed.

Immunologic Drug-Induced Thrombocytopenia
Many medications can result in severe thrombocytopenia. Depending on the medicine and platelet activity of the antibody, drug-induced immune-mediated thrombocytopenia can be divided into several categories.
Drug-dependent antibodies
One mechanism of drug-dependent antibodies is quinidine and quinine-induced thrombocytopenia. Even when the treatment is active, the antibody produced by these drugs binds to platelets. Opioid-based antibodies often form 1 to 2 weeks following the administration of a new medicine. Many medicines can cause such antibodies, although quinine, quinidine, and sulfonamide compounds do so more commonly than others. When antibody formation begins, the platelet count decreases dramatically and can never go above 10,000/μL. Patients may have bleeding symptoms abruptly. If a pregnant woman develops this type of thrombocytopenia as a result of the medicine, it may have an impact on both the fetus and the mother.
Common Drugs Causing Immune Thrombocytopenia
| Analgesics | Salicylates Acetaminophen Phenylbutazone |
| Antibiotics | Cephalothin Penicillin Streptomycin Aminosalicylic acid Rifampin Novobiocin Various sulfa drugs (chlorthalidone, furosemide) |
| Alkaloids | Quinidine Quinine |
| Sedatives, Anticonvulsants | Methoin Troxidone Chlorpromazine Diphenylhydantoin Meprobamate Phenobarbital Carbamazepine |
| Oral Hypoglycemics | Chlorpropamide Tolbutamide |
| Heavy Metals | Gold Mercury Bismuth Organic arsenicals |
| Miscellaneous | Chloroquine Chlorothiazide Insecticides |
Hapten-induced antibodies
A second type of thrombocytopenia induced by the medication is the production of hapten-dependent antibodies. Any drug molecule is too small to produce an immune response on its own, but it may operate as a hapten and bind with a bigger carrier molecule (often a platelet membrane plasma protein or protein component) to form a complex that can serve as a complete antigen. The medication-induced thrombocytopenia is almost always threatening. The initial platelet count can be less than 10,000/μL, and in certain cases less than 1,000/μL. The presence of megakaryocytes in bone marrow is typically normal to increased.
Drug-induced autoantibodies
A third cause of thrombocytopenia caused by medicine is drug-mediated autoantibodies. In this case, the medications promote the development of autoantibodies that bind to a specific platelet membrane glycoprotein and without any requirement for the presence of free drugs. Gold salts and procainamide are 2 type of these drugs.
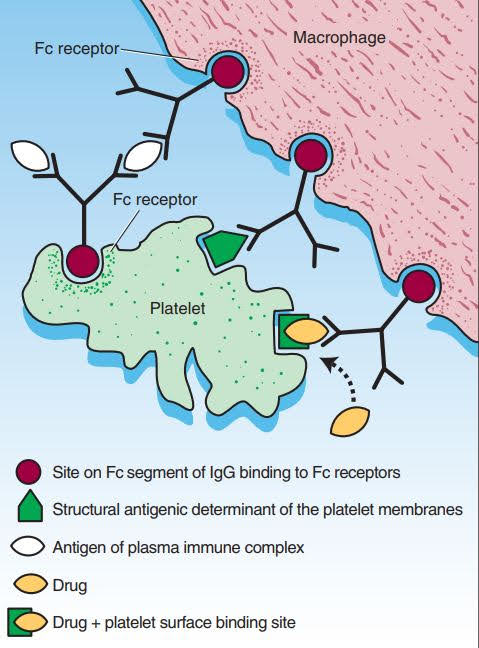
Immune complex–induced thrombocytopenia
Another kind of thrombocytopenia caused by the medication is heparinduced thrombocytopenia (HIT). Platelet factor 4 (PF4), a protein formed and secreted by platelets that neutralizes heparin, binds to heparin. Heparin binding by PF4 plasma or PF4 platelet membrane-expressed PF4 causes a conformational change in PF4, leading in neoepitope expression. The exposure to such neoepitopes (“new antigens”) triggers the immune response of certain people, contributing to the development of an antibody at one of the neoepitopes. Heparin and PF4 combine to create a combination on the platelet surface or circulating free complexes to which the antibody attaches during HIT. The immunoglobulin molecule’s Fab part attaches to an exposed neoepitope in the PF4 molecule, allowing the Fc portion of the IgG to bind.
The role of Helicobacter pylori in the development of ITP
Following bacterial treatment, platelet counts in ITP patients positive for H. Pylori improved in a number of studies. However, the results of several tests are inconsistent, with some facilities having a very great reaction rate to treatment and prevention and others having a minimal response rate. H. Pylori role in ITP resulted from investigations suggest that the number of antiplatelet antibodies in plasma decreases following bacterial treatment.
T cells may also be involved
The involvement of T cells in ITP development is becoming obvious. Because the autoantibodies in ITP are mostly of the IgG subclass, T cells have been associated for such, and isotype change from IgM to IgG requires T-cell assistance. However, over 40% of chronic ITP patients have no detectable autoantibodies, are thrombocytopenic, and appear to have true ITP. CD8 + T cells have been linked to the development of several autoimmune illnesses, including type 1 diabetes, and it has recently been demonstrated that ITP patients have a significant CD8 + T cell-a driven cytotoxicity that causes platelet loss. However, it is uncertain whether cell-mediated platelet degradation adds to disease severity or not.
References:
- Rodaks Hematology 5th Edition
- Post Graduate Hematology 6th Edition