
Table of Contents
Spinal muscular atrophy (SMA) is a rare autosomal recessive neuromuscular disorder characterized by the loss of lower motor neurons (anterior horn cells) in the spinal cord and brainstem nuclei, resulting in progressive muscle weakening and atrophy. The severity and age at onset differ considerably.
SMA was initially documented in the 1890s by Guido Werdnig of the University of Vienna, who diagnosed intermediate and severe SMA in two individuals with developing lower extremity weakness. The name “Spinale Muskelatrophie” was coined by Johan Hoffmann of Hiedelberg University. Kugelberg and Welander also reported milder types of SMA. Byers and Banker first categorized the SMA into three groups:
- Group 1: Intrauterine presentation or clinical symptoms in the first two postnatal months, marked by early weakness and mortality.
- Group 2: Initial onset between the ages of 2 and 12 months, with more localized weakness and prolonged survival.
- Group 3: Presentation after 1 year of age
How is the SMA caused?
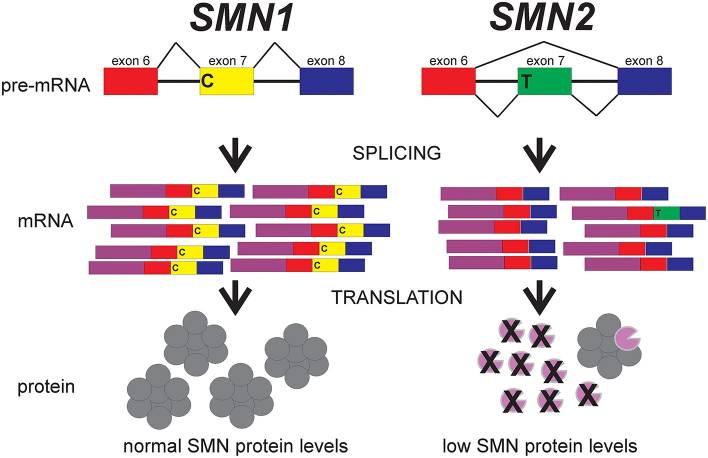
Although SMA is caused by a homozygous deletion of SMN1 on chromosome 5q13 in 95 percent of patients, this does not explain why there’s considerable clinical variation in phenotype. The evidence is that there are two types of SMN: telemoeric (SMN1) and centromeric (SMN2), with individuals differing in the number of copies of SMN2. SMN1 transcription generates a functionally complete mRNA, which can subsequently be translated into the survival motor neuron (SMN) protein. Because SMN2 transcription produces functionally full mRNA just 10 to 15% of own overall, remarkably fewer SMN proteins are produced than SMN1.
Except for a single C-T polymorphism in exon 7, SMN2 is identical to SMN1. This variation induces splicing 80 to 85 % during transcription, resulting in the loss of exon 7. This mRNA truncation results in similarly shortened non-functional proteins. Patients with SMA lack SMN1 and must rely on residual SMN2 synthesis of functional SMN protein for alpha motor neuron function and survival. As a result, there is a positive association between the number of SMN2 copies and phenotypic severity, with SMA type 1 often having 1 to 2 copies of SMN2 and SMA type 4 typically having 3 to 5 SMN2 copies.
Even though SMA has been reported by the prevalence of SMN1 deletions and mutations, clinical presentation varies depending on the existence of an adjoining and almost identical genes, SMN2. Both the SMN1 and SMN2 genes can generate the full-length SMN mRNA transcript necessary for normal SMN protein production, as well as other less consistent transcripts. While the full-length transcript is the main product of SMN1, SMN2 generates less of the full-length SMN mRNA transcript and thus less than the full-length (functional) SMN protein.
In general, the more copies of SMN2 present in a SMA patient, the milder the symptoms. The loss of SMN1 function would be fatal without SMN2 copies, but individuals with 5 or more copies of SMN2 may have no symptoms at all. However, even among family members, SMN2 copy quantity does not perfectly correspond with phenotype.
Types of Spinal Muscular Atrophy
SMA is now classified based on clinical symptoms and greatest functional accomplishment. SMA type 0 is perhaps the most severe kind, causing weakness in prenatal or at births.
- SMA TYPE 0
- Typical Age at onset: Prenatal/birth
- Typical number of SMN2 copies: 0-1
- Frequency: <5%
- Maximum motor function: It manifests during the prenatal and neonatal periods as a lack of fetal movement, contractures, and severe hypotonia. Death occurs early in the absence of intensive supportive care starting at delivery or shortly thereafter.
- Life Expectancy: Neonatal period
- SMA TYPE 1
- Typical Age at onset: 0-6 months
- Typical number of SMN2 copies: 1,2,3
- Frequency: ~60%
- Maximum motor function: Werdnig-Hoffman disease; Never sits independently.
- Life Expectancy: <2 years
- SMA TYPE 2
- Typical Age at onset: 6-18 months
- Typical number of SMN2 copies: 2,3,4
- Frequency: ~10%
- Maximum motor function: Dubowitz disease; Capable of sitting but unable to walk independently
- Life Expectancy: >2 years
- SMA TYPE 3
- Typical Age at onset: >18 months
- Typical number of SMN2 copies: 3,4
- Frequency: <5%
- Maximum motor function: Kugelberg-Welander disease; Shows significant diversity in onset, symptom development, and function, but can eventually stand or walk independently.
- Life Expectancy: Normal life expectancy
- SMA TYPE 4
- Typical Age at onset: >21 years
- Typical number of SMN2 copies: > or = 4
- Frequency: ~20%
- Maximum motor function: Presents in adulthood. Life expectancy is normal. Individuals with more than 6 copies may be phenotypically normal.
- Life Expectancy: >2 years
Genetics of SMA
The majority of SMA cases are associated with mutations in the SMN1 gene, which is located at 5q13.12. SMN1 (also known as SMNT, where T stands for telomere) spans 20kb and is located in the telomeric part of a 500kb inverted duplication, which is susceptible to rearrangements and deletions. SMN2 (also known as C-BCD541 and SMNC, where C stands for centromere) is a highly homologous centromeric element that has more than 99 percent nucleotide similarity with SMN1. Both SMN1 and SMN2 have 9 exons that encode the 294-amino acid protein, motor neuron survival (SMN). Exons are labeled 1, 2, 2a, 2b, 3, 4, 5, 6, 7, and 8.
Exon 7 contains the SMN stop codon, while exon 8 remains untranslated. The differences between SMN1 and SMN2 are 8 nucleotides, 5 of which are intronic and 3 of which are in the final three exons. Only one change (c.840C>T) in SMN2 exon 7 lies in a coding region and disables an exonic splice enhancer in exon 7. As a result of this mutation, the majority of SMN2 transcripts miss exon 7, resulting in an incomplete and degraded SMN protein. Each SMN2 copy produces an estimated 10% of functional protein, making it a modifying gene. SMN is an RNA-binding protein that is involved in a variety of biological activities and pathways, most notably in the snRNP complex.
The major functional difference between these SMN1 and SMN2 genes is a C-to-T transition in exon 7 (SMN2 c.850C>T). While translationally silent, this site on exon 7 is in the midst of an exonic splicing enhancer (ESE) region required for exon 7 inclusion in SMN transcripts. This ESE is broken in SMN2, resulting in the exclusion of exon 7 (SMNΔ7) from the bulk (90%) of SMN2-derived mRNAs. The resulting SMNΔ7 protein is unstable and unable to connect with itself. Depending on the cell type, certain SMN2 mRNAs have exon 7 and can create some full-length, functional SMN proteins.
Pathophysiology
SMN is a ubiquitously produced protein with several functions in cells, and its expression is lowered in SMA. The SMN protein is a 38-kDa protein that is found in the cytoplasm and nucleus of all tissues, with the highest concentrations in the brain, spinal cord, and muscle. SMN has been linked to RNA metabolism and processing. Two important assumptions attribute to SMN protein’s function in 1) The neuronal cytoplasm and 2) the neuronal nucleus.
SMN is found in nuclear structures known as gemini of coiled bodies, or gems, and is hypothesized to have a role in pre-mRNA splicing and other RNA metabolic processes. The SMN complex is involved in the cytoplasmic production of uridine-rich small nuclear ribonucleoproteins (snRNP), which are materials of both the main and minor spliceosomes.
The SMN complex is necessary for Sm protein assembly as well as directed recruitment of snRNA, which is essential for snRNP integrity and functionality. As a result, decreased SMN levels result in a loss of spliceosomal snRNPs. This interpretation explains the destruction to motor neurons to either neuronal sensitivity to spliceosome dysfunction directly or indirectly through improperly spliced mRNA producing dysfunctional proteins critical to neuronal function.
Despite the early idea that SMA is only a motor neuron condition, data from thoroughly phenotypically examined SMA mice, together with investigations in SMA humans, shows that most organs are affected. Importantly, significantly lowered SMN levels have a direct effect on cell-specific pathways in the majority of such non-neuronal cells and tissues without becoming related to Motor Neuron dysfunction.
SMN1 and SMN2 Copy Number
As well, since SMN2 copy number increases disease severity in SMA, reliable quantification of SMN2 copy number in SMA patients has prognostic value. The count of SMN2 copies in the genome ranges between 0 and 6. Due to the sheer instability of the 5q13.2 locus, the copy number of both SMN1 and SMN2 is extremely variable, resulting in a high incidence of deletion/duplication and gene conversion events. Numerous studies have found an inverse association between SMN2 copy number and disease severity in SMA patients. The increased SMN2 copy in SMA is attributed to gene conversion from SMN1 to SMN2. However, SMN2 copy number is not a definite variable of SMA severity, and several influencing parameters must be considered for disease characterization.

Since then, several methods for quantifying SMN2 copy number in DNA samples from SMA patients have been developed. Radioactive PCR fluorescent PCR, quantitative (real-time) PCR, competitive PCR/primer extension, denaturing high performance liquid chromatography, multiplex ligation-dependent probe amplification, quantitative capillary electrophoresis fragment analysis, short-amplicon melt profiling, fluorescent multiplex PCR/capillary electrophoresis, and universal fluorescent tri-probe ligation are some of the assays available. dPCR is capable of measuring SMN1 and SMN2 copy counts across a huge spectrum, i.e., between 0 and 6 copies.
Referencs:
- Keinath MC, Prior DE, Prior TW. Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. Appl Clin Genet. 2021 Jan 25;14:11-25. doi: 10.2147/TACG.S239603. PMID: 33531827; PMCID: PMC7846873.
- Ross LF, Kwon JM. Spinal Muscular Atrophy: Past, Present, and Future. Neoreviews. 2019 Aug;20(8):e437-e451. doi: 10.1542/neo.20-8-e437. PMID: 31371553.
- Burr P, Reddivari AKR. Spinal Muscle Atrophy. 2021 Aug 11. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 32809522.
- Butchbach MER. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. Int J Mol Sci. 2021 Jul 23;22(15):7896. doi: 10.3390/ijms22157896. PMID: 34360669; PMCID: PMC8348669.
- Butchbach ME. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front Mol Biosci. 2016 Mar 10;3:7. doi: 10.3389/fmolb.2016.00007. PMID: 27014701; PMCID: PMC4785180.
- Arnold ES, Fischbeck KH. Spinal muscular atrophy. Handb Clin Neurol. 2018;148:591-601. doi: 10.1016/B978-0-444-64076-5.00038-7. PMID: 29478602.
- Nash LA, Burns JK, Chardon JW, Kothary R, Parks RJ. Spinal Muscular Atrophy: More than a Disease of Motor Neurons? Curr Mol Med. 2016;16(9):779-792. doi: 10.2174/1566524016666161128113338. PMID: 27894243.
- Farrar MA, Kiernan MC. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics. 2015 Apr;12(2):290-302. doi: 10.1007/s13311-014-0314-x. PMID: 25413156; PMCID: PMC4404441.
- Wirth B. Spinal Muscular Atrophy: In the Challenge Lies a Solution. Trends Neurosci. 2021 Apr;44(4):306-322. doi: 10.1016/j.tins.2020.11.009. Epub 2021 Jan 7. PMID: 33423791.
- Vijzelaar R, Snetselaar R, Clausen M, Mason AG, Rinsma M, Zegers M, Molleman N, Boschloo R, Yilmaz R, Kuilboer R, Lens S, Sulchan S, Schouten J. The frequency of SMN gene variants lacking exon 7 and 8 is highly population dependent. PLoS One. 2019 Jul 24;14(7):e0220211. doi: 10.1371/journal.pone.0220211. PMID: 31339938; PMCID: PMC6655720.